cellNexus is a query interface for programmatic exploration and retrieval of harmonised, curated, and reannotated CELLxGENE human-cell-atlas data.
This standalone documentation website provides:
- Detailed data-processing information (quality control, harmonisation, and expression representations).
- Complete explanations of metadata columns used in filtering and interpretation.
- Guided examples for metadata-first exploration and gene-expression analysis.
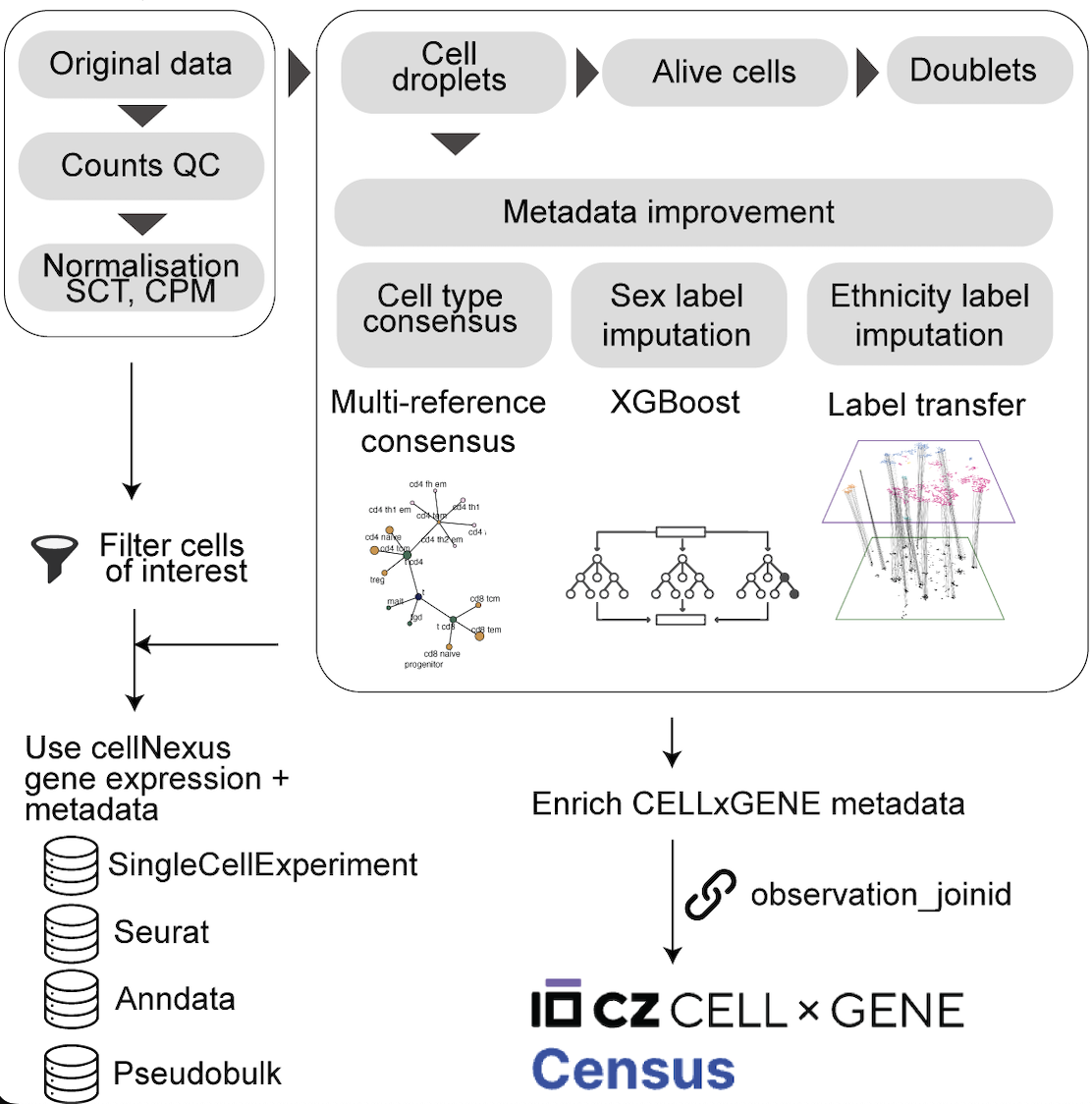
Data Processing Overview
The harmonisation pipeline standardises data across datasets so queries are consistent across studies:
- Metadata are retrieved from cloud-hosted harmonised tables.
- Standardised quality control removes empty droplets, dead/damaged cells, and likely doublets.
- Cell-level data are served through common assay layers (
counts,cpm,sct,pseudobulk). - Outputs are returned in analysis-ready R formats such as
SingleCellExperimentandSeurat.
Quality control steps
The QC flags used throughout cellNexus are computed on sample-level using HPCell. In brief:
- Empty droplets (
empty_droplet):- Excludes mitochondrial genes and ribosomal genes before scoring.
- Computes, per cell, the number of expressed genes and flags a cell as an empty droplet when the number of expressed genes is less than
RNA_feature_threshold(by default 200, except for targeted panels such as Rhapsody technology).
- Alive cells (
alive):- Filters out empty droplets.
- Computes per-cell QC metrics from raw counts using
scuttle::perCellQCMetrics(..., subsets=list(Mito=...)), where the mitochondrial subset is defined by^MTin the feature names. - Determines high mitochondrial content via
scater::isOutlier(subsets_Mito_percent, type="higher"). Outlier calling is performed within each cell-type group using our harmonised labelcell_type_unified_ensemble. - Alive cells are labelled as those without high mitochondrial content (
!high_mitochondrion).
- Doublets (
scDblFinder.class):- Filters out empty droplets.
-
scDblFinder::scDblFinder()default parameters are used. For cells that cannot be classified byscDblFinder, the label is set to"Unknown"to avoid dropping cells.
RNA abundance
- RNA counts:
- RNA count distributions per sample are annotated from cellxgenedp, using the
x_approximate_distributioncolumn.
- RNA count distributions per sample are annotated from cellxgenedp, using the
- CPM:
- Counts-per-million normalisation computed from the raw counts assay via
scuttle::calculateCPM().
- Counts-per-million normalisation computed from the raw counts assay via
- Rank:
- Per-cell gene-expression ranks computed with
singscore::rankGenes(). - Implemented in column chunks (default 1000 cells per slice) to handle very large datasets; slices are written to disk as an
HDF5Array-backed sparse integer matrix and then column-bound.
- Per-cell gene-expression ranks computed with
- SCT:
- All low-quality cells flagged by QC are removed before normalisation.
- Variance-stabilising normalisation computed with Seurat
SCTransform()(v2), with regression of cell-level covariates (subsets_Mito_percentandsubsets_Ribo_percent). - The median common scale across the whole resource is applied (scale_factor=2186)
- Pseudobulk:
- All low-quality cells flagged by QC are removed before aggregation.
- Aggregates counts across cells using
scuttle::aggregateAcrossCells(), aggregatingsample_idand the harmonised cell type (cell_type_harmonised_ensemble).
Metadata Explore
Through harmonisation and curation, cellNexus adds columns that are not present in the original CELLxGENE metadata alone. The latest metadata is downloaded automatically when you call get_metadata() — no need to supply a download link manually. See the client usage examples for details on the columns available.
| Column | Description |
|---|---|
cell_id |
Cell identifier. |
observation_joinid |
Cell ID join key linking metadata. |
dataset_id |
Primary dataset identifier in the atlas. |
sample_id |
Harmonised sample identifier. |
sample_ |
Internal sample subdivision helper. |
sample_heuristic |
Internal sample subdivision helper. |
age_days |
Donor age in days. |
tissue_groups |
Coarse tissue grouping for analysis. |
nFeature_expressed_in_sample |
Number of expressed features per cell. |
nCount_RNA |
Total RNA counts per cell (sample-aware). |
empty_droplet |
Quality-control flag for empty droplets. |
cell_type_unified_ensemble |
Consensus immune identity from Azimuth and SingleR (Blueprint, Monaco). |
is_immune |
Curated flag for immune-cell context. |
subsets_Mito_percent |
Percent of each cell’s total counts coming from mitochondrial genes in a sample. |
subsets_Ribo_percent |
Percent of each cell’s total counts coming from ribosomal genes in a sample. |
high_mitochondrion |
TRUE if the cell’s mitochondrial percent exceeds the QC cutoff. |
high_ribosome |
TRUE if the cell’s ribosomal percent exceeds the QC cutoff. |
scDblFinder.class |
Quality-control flag for doublet classification from scDblFinder. |
sample_chunk |
Internal sample subdivision chunks. |
cell_chunk |
Internal cell subdivision chunks. |
sample_pseudobulk_chunk |
Internal pseudobulk subdivision chunks. |
file_id_cellNexus_single_cell |
Internal file id for single-cell layers. |
file_id_cellNexus_pseudobulk |
Internal file id for pseudobulk layers. |
count_upper_bound |
Count capping threshold used in transformation. |
nfeature_expressed_thresh |
Threshold of the number of expressed features per cell. |
inverse_transform |
Transformation method used in pre-processing pipeline. |
alive |
Quality-control flag for viable cells (e.g. mitochondrial signal). |
cell_annotation_blueprint_singler |
SingleR annotation (Blueprint). |
cell_annotation_monaco_singler |
SingleR annotation (Monaco). |
cell_annotation_azimuth_l2 |
Azimuth cell annotation. |
ethnicity_flagging_score |
Supporting score for ethnicity imputation. |
low_confidence_ethnicity |
Supporting flag for low-confidence ethnicity calls. |
.aggregated_cells |
Post-QC cells combined into each pseudobulk sample. |
imputed_ethnicity |
Imputed ethnicity label. |
atlas_id |
cellNexus atlas release identifier (internal use). |
Field definitions for the CELLxGENE schema follow the CELLxGENE schema 5.1.0, and CELLxGENE Census schema
Client Usage Examples
R client (cellNexus)
library(cellNexus)
library(dplyr)
library(stringr)
metadata <- get_metadata()
metadata <- metadata |>
keep_quality_cells()
census_metadata <- cellNexus:::get_census_metadata("2024-07-01")
con <- dbplyr::remote_con(metadata)
duckdb::duckdb_register_arrow(con, "census_metadata", census_metadata)
metadata <- metadata |>
dplyr::left_join(tbl(con, "census_metadata"))
query <- metadata |>
filter(
self_reported_ethnicity == "African",
str_like(assay, "%10x%"),
tissue == "lung parenchyma",
str_like(cell_type, "%CD4%")
)
sce <- get_single_cell_experiment(query, assays = c("counts", "cpm"))
pb <- get_pseudobulk(query)Python client (cellNexusPy)
Python support is available in the companion repository: MangiolaLaboratory/cellNexusPy.
from cellnexuspy import get_metadata, join_census_table, keep_quality_cells, get_anndata, get_pseudobulk
conn, table = get_metadata()
metadata = join_census_table(conn, table)
metadata = keep_quality_cells(metadata)
query = metadata.filter("""
self_reported_ethnicity == 'African' AND
assay LIKE '%10x%' AND
tissue == 'lung parenchyma' AND
cell_type LIKE '%CD4%'
""")
sce = get_anndata(query, assays=["counts","cpm"])
pb = get_pseudobulk(query)
conn.close()For other implementation details and code examples, see cellNexus README